GWAS로 배우는 유전통계학 - 1. 시작하며
2012-03-30
Genome-wide association analysis
GWAS
유전통계학
이제 전 게놈 관련분석은 어느 정도 정형화된 분석방법이 아닌가 싶습니다. 예전부터 이 분석방법에 대해 한번 정리해보고 싶었는데, 마침 일본 계산기통계학회에서 종합보고서 형식의 글을 써달라는 제의가 왔기에 회사 동료와 함께 작성한 글을 바탕으로 앞으로 5~6회에 걸쳐 GWAS에 대해 정리해 보고자 합니다.
1. 시작하며
인간의 다양성(variation)에 대한 해명은 근대 통계학연구의 좋은 재료였습니다. 앞선 포스팅(유전통계학과 수리통계학의 역사)에서 자세히 살펴본 바와 같이 Galton, K. Pearson, Fisher로 대표되는 통계학자들은 인간의 다양성을 설명하기 위해 회귀분석, 검정, 우도 등 근대통계학의 기초를 세우게 됩니다. 특히 Fisher는 멘델의 법칙에 따라 안정된 상태로 다음 세대로 다양한 정보가 전달되며 그 결과로써 각종 다양성이 발생한다고 생각하였습니다. 이처럼 멘델의 법칙에 따라 다양성이 만들어진다고 생각한 연구그룹을 멘델학파(medelian)라 합니다. 그러나 당시에는 게놈정보의 관측이 어려웠으므로 관측 데이터로부터 다양성의 메커니즘을 평가하는 생물계측학파(biometrician)가 실세를 차지하고 있었습니다.
하지만 1970년대에 들어 Sanger에 의해 획기적인 게놈정보 관측기술이 개발되고 게놈정보의 관측이 가능해져 멘델학파는 연관분석(linkage analysis)이라는 방법으로 유전적 요인이 비교적 강한 질병의 원인 유전자를 찾아내는 데 성공합니다. 또한, 21세기에 들어서 DNA chip이 출현하게 되고 모든 염색체에 존재하는 게놈정보를 싼 가격에 관측할 수 있게 되어, 지금까지 특수한 연구분야였던 게놈연구가 일반적으로 이루어지게 되었습니다.
전 게놈 관련분석(Genome-wide association study; GWAS)은 병질환 및 약물 반응성에 대한 유전적 요인을 총체적으로 탐색하는 연구 방법을 말하며, 일본 이화학연구소의 Ozaki(2002) 그룹에서 최초로 시도된 연구 방법입니다. 이후 게놈정보의 관측기술 및 분석기술의 발전 덕분에 세계 각지에서 많은 수의 연구 결과가 보고되고 있으며, 최근, 특히 올해 들어서 NGS(Next generation sequencing)을 이용한 논문 수가 급격하게 늘고 있기는 하지만 아직 Nature Genetics 등 주요 저널에 실리는 논문의 약 절반가량이 GWAS 연구 결과로 채워지고 있습니다. GWAS의 연구 성과에 대해서는 National Human Genome Research Institute의 GWAS catalog에 정리되어 있으니 참고하길 바랍니다.
형질의 다양성이 유전자 다형성에 그 원인이 있다는 전제를 바탕으로 GWAS는 질환의 유무, 약물 복용에 따른 부작용의 유무 등과 같은 질적 형질(Qualitative trait)및 혈액 검사치 등의 양적 형질(Quantitative trait)과 DNA에 존재하는 유전자 다형성(polymorphism)을 그 분석 대상으로 합니다. 다양성의 기초가 되는 유전자 다형성은 인간의 약 30억 염기배열쌍(염기 A, T, G, C의 배열) 중 약 1%정도 존재하며 다형성을 표현하기 위한 다양한 지표가 존재함은 앞선 글(게놈의 다양성과 유전자 다형성)에서 설명한 바와 같습니다.
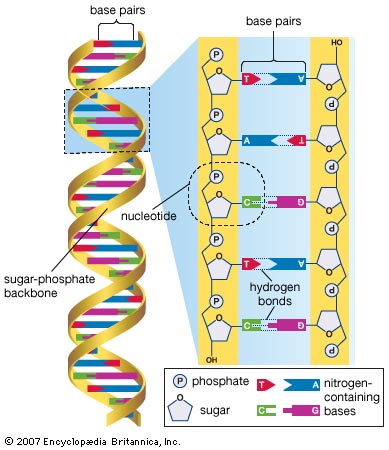
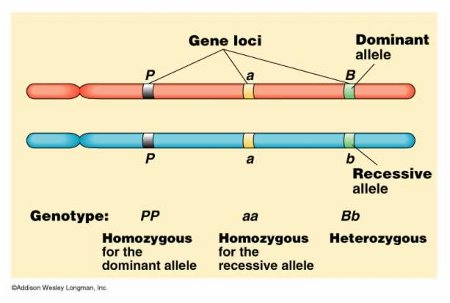
그중에서도 GWAS는 유전자 다형성의 하나인 단염기치환(Single nucleotide polymorphism; SNP)를 주로 이용합니다. 이 SNP가 존재하는 장소를 SNP좌위(SNP locus)라고 부르며 대개의 경우 하나의 SNP좌위는 두 염기의 조합으로 이루어집니다. 인간은 부모로부터 각각 하나의 염기를 물려받기 때문에 이 두 염기의 조합으로 유전적 요인을 표현할 수 있으며 이를 유전자형(genotype)이라 합니다. 예를 들어 염기 A와 T로 구성된 SNP에서는 개인에 따라 AA, AT, TT 중 하나의 유전자형을 가지게 됩니다. 또한, 염기와 같이 안정된 형태로 다음 세대에 전달되는 대상물을 보다 광범위하게 대립유전자(allele, 이후 allele로 표기)라 합니다. 앞선 예의 염기 A, T가 바로 각각의 allele에 해당하며 유전자형은 2개의 allele의 조합이라 할 수 있습니다. 따라서 유전 좌위(locus, 복수형은 loci)는 allele이 존재하는 장소를 일컫는 용어이기도 합니다.
대량의 SNP좌위의 유전자형 관측에는 DNA chip을 이용합니다. GWAS는 게놈 전부를 탐색한다는 의미의 Genome-wide라는 표현을 사용하고 있지만 실제로는 인간의 약 30억 염기쌍을 분석하는 것이 아니라 미리 DNA chip에 탑재된 50만~250만 SNP좌위의 유전자형을 관측하게 됩니다. 10년 전만 해도 하나의 SNP를 관측하기 위해 한 사람당 약 1달러 정도의 비용이 들었지만, DNA chip의 등장 덕분에 현재는 SNP당 약 0.05센트 정도의 비용으로 관측할 수 있게 되었습니다. DNA chip의 종류에 따라 관측좌위 수 및 부위가 다르므로 연구의 목적에 따라 선택해야 합니다.
GWAS에서 통계분석의 주목적은 추정과 검정이라 할 수 있습니다. GWAS에서는 주로 병에 걸린 사람과 정상인에 대해 SNP좌위의 유전자형 빈도 차이에 대해 오즈비(odds ratio)를 추정하거나 다량의 SNP좌위에 대해 분할표의 검정을 하게됩니다. 비교 대상이 되는 집단의 환경적 요인에 차이가 있다면 통계모형을 도입하여 SNP좌위 이외에 연령, 성별 등 환경요인을 설명변수로 하는 회귀모형을 이용하기도 합니다. 그 이외에도 생존시간분석, 기계학습모형의 도입 등 다양한 분석이 이루어지고 있습니다.
GWAS를 시작으로 하는 데이터 분석에서 게놈정보를 다룰 때 절대적인 규칙이 있으니 그것이 바로 물리적으로 관측 가능한 법칙인 유전계승법칙(law of inheritance)입니다. 여기서 유전계승법칙이라 함은 멘델의 세 가지 법칙, “분리의 법칙”, “독립의 법칙”, “우열의 법칙”과 독립 법칙의 예외인 연쇄(linkage)의 법칙을 말합니다. 게놈분석에서는 데이터의 관측으로부터 분석에 이르기까지 모든 장면에서 유전계승법칙을 무시할 수 없습니다. 특히 GWAS의 분석대상이 되는 SNP좌위는 그 양이 매우 많으므로 통계학에서의 제1종의 오류(Type 1 error)도 빈번히 일어납니다. 때문에 제1종의 오류를 줄이기 위한 다중비교 문제는 GWAS에서 피할 수 없는 문제 중 하나입니다. 또한, DNA chip의 관측오류도 무작위적으로 일어나는 것이 아니라 일정한 경향성이 있는 것도 알려져 있습니다. 이 때문에 조금이라도 거짓 양성을 줄이기 위해서는 참된 모형인 유전계승법칙에 근거해 분석을 진행하는 것이 자연스럽다 할 수 있겠습니다.
GWAS를 위한 도입설명은 이쯤 하기로 하고 다음 포스팅에서는 실제 분석을 진행하기 전에 필요한 데이터의 품질평가 방법들에 대해 적어 보도록 하겠습니다.
참고문헌
- Balding D.J. (2006), Nature reviews Genetics, 7, 10, 781-791.
- Kruglyak L. (2008), Nature reviews Genetics, 9, 4, 314-318.
- Ozaki K et al (2002), Nature Genetics, 32, 4, 650-654.
- 鎌谷直之 (2007) 遺伝統計学入門, 岩波書店 (카마타니 나오유키 (2007), 유전통계학 입문, 이와나미서점 )